Nextflow Development - Creating a Nextflow Workflow
Nextflow Channels and Processes
- Gain an understanding of Nextflow channels and processes
- Gain an understanding of Nextflow syntax
- Read data of different types into a Nextflow workflow
- Create Nextflow processes consisting of multiple scripting languages
3.1.1. Environment Setup
Clone the training materials repository on GitHub:
git clone https://github.com/nextflow-io/training.gitSet up an interactive shell to run our Nextflow workflow:
srun --pty -p prod_short --mem 8GB --mincpus 2 -t 0-2:00 bashLoad the required modules to run Nextflow:
module load nextflow/23.04.1
module load singularity/3.7.3Make sure to always use version 23 and above, as we have encountered problems running nf-core workflows with older versions.
Since we are using a shared storage, we should consider including common shared paths to where software is stored. These variables can be accessed using the NXF_SINGULARITY_CACHEDIR or the NXF_CONDA_CACHEDIR environment variables.
Currently we set the singularity cache environment variable:
export NXF_SINGULARITY_CACHEDIR=/config/binaries/singularity/containers_devel/nextflowSingularity images downloaded by workflow executions will now be stored in this directory.
You may want to include these, or other environmental variables, in your .bashrc file (or alternate) that is loaded when you log in so you don’t need to export variables every session. A complete list of environment variables can be found here.
3.1.2. Nextflow Workflow
A workflow can be defined as sequence of steps through which computational tasks are chained together. Steps may be dependent on other tasks to complete, or they can be run in parallel.
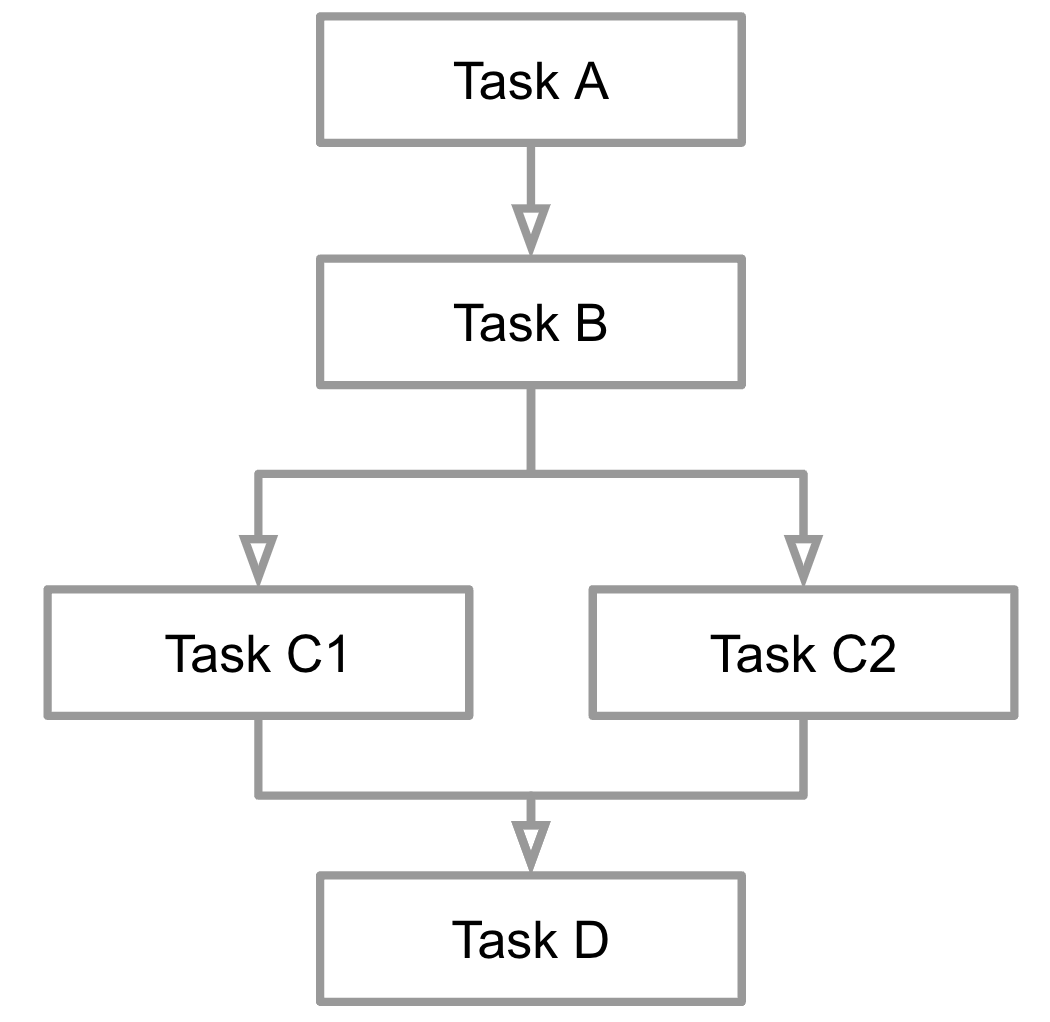
In Nextflow, each step that will execute a single computational task is known as a process. Channels are used to join processes, and pass the outputs from one task into another task.
3.1.3. Channels and Channel Factories
Channels are a key data structure of Nextflow, used to pass data between processes.
Queue Channels
A queue channel connects two processes or operators, and is implicitly created by process outputs, or using channel factories such as Channel.of or Channel.fromPath.
The training/nf-training/snippet.nf script creates a channel where each element in the channel is an arguments provided to it. This script uses the Channel.of channel factory, which creates a channel from parameters such as strings or integers.
ch = Channel.of(1, 2, 3)
ch.view()The following will be returned:
>>> nextflow run training/nf-training/snippet.nf
N E X T F L O W ~ version 23.04.1
Launching `training/nf-training/snippet.nf` [shrivelled_brattain] DSL2 - revision: 7e2661e10b
1
2
3Value Channels
A value channel differs from a queue channel in that it is bound to a single value, and it can be read unlimited times without consuming its contents. To see the difference between value and queue channels, you can modify training/nf-training/snippet.nf to the following:
ch1 = Channel.of(1, 2, 3)
ch2 = Channel.of(1)
process SUM {
input:
val x
val y
output:
stdout
script:
"""
echo \$(($x+$y))
"""
}
workflow {
SUM(ch1, ch2).view()
}This workflow creates two queue channels, ch1 and ch2, that are input into the SUM process. The SUM process sums the two inputs and prints the result to the standard output using the view() channel operator.
After running the script, the only output is 2, as below:
>>> nextflow run training/nf-training/snippet.nf
N E X T F L O W ~ version 23.04.1
Launching `training/nf-training/snippet.nf` [modest_pike] DSL2 - revision: 7e2661e10b
2Since ch1 and ch2 are queue channels, the single element of ch2 has been consumed when it was initially passed to the SUM process with the first element of ch1. Even though there are other elements to be consumed in ch1, no new process instances will be launched. This is because a process waits until it receives an input value from all the channels declared as an input. The channel values are consumed serially one after another and the first empty channel causes the process execution to stop, even though there are values in other channels.
To use the single element in ch2 multiple times, you can use the Channel.value channel factory. Modify the second line of training/nf-training/snippet.nf to the following: ch2 = Channel.value(1) and run the script.
>>> nextflow run training/nf-training/snippet.nf
N E X T F L O W ~ version 23.04.1
Launching `training/nf-training/snippet.nf` [jolly_archimedes] DSL2 - revision: 7e2661e10b
2
3
4Now that ch2 has been read in as a value channel, its value can be read unlimited times without consuming its contents.
In many situations, Nextflow will implicitly convert variables to value channels when they are used in a process invocation. When a process is invoked using a workflow parameter, it is automatically cast into a value channel. Modify the invocation of the SUM process to the following: SUM(ch1, 1).view() and run the script”
>>> nextflow run training/nf-training/snippet.nf
N E X T F L O W ~ version 23.04.1
Launching `training/nf-training/snippet.nf` [jolly_archimedes] DSL2 - revision: 7e2661e10b
2
3
43.1.4. Processes
In Nextflow, a process is the basic computing task to execute functions (i.e., custom scripts or tools).
The process definition starts with the keyword process, followed by the process name, commly written in upper case by convention, and finally the process body delimited by curly brackets.
The process body can contain many definition blocks:
process < name > {
[ directives ]
input:
< process inputs >
output:
< process outputs >
[script|shell|exec]:
"""
< user script to be executed >
"""
}- Directives are optional declarations of settings such as
cpus,time,executor,container. - Input defines the expected names and qualifiers of variables into the process
- Output defines the expected names and qualifiers of variables output from the process
- Script is a string statement that defines the command to be executed by the process
Inside the script block, all $ characters need to be escaped with a \. This is true for both referencing Bash variables created inside the script block (ie. echo \$z) as well as performing commands (ie. echo \$(($x+$y))), but not when referencing Nextflow variables (ie. $x+$y).
process SUM {
debug true
input:
val x
val y
output:
stdout
script:
"""
z='SUM'
echo \$z
echo \$(($x+$y))
"""
}By default, the process command is interpreted as a Bash script. However, any other scripting language can be used by simply starting the script with the corresponding Shebang declaration. To reference Python variables created inside the Python script, no $ is required. For example:
process PYSTUFF {
debug true
script:
"""
#!/usr/bin/env python
x = 'Hello'
y = 'world!'
print ("%s - %s" % (x, y))
"""
}
workflow {
PYSTUFF()
}Vals
The val qualifier allows any data type to be received as input. In the example below, num queue channel is created from integers 1, 2 and 3, and input into the BASICEXAMPLE process, where it is declared with the qualifier val and assigned to the variable x. Within this process, the channel input is referred to and accessed locally by the specified variable name x, prepended with $.
num = Channel.of(1, 2, 3)
process BASICEXAMPLE {
debug true
input:
val x
script:
"""
echo process job $x
"""
}
workflow {
BASICEXAMPLE(num)
}In the above example the process is executed three times, for each element in the channel num. Thus, it results in an output similar to the one shown below:
process job 1
process job 2
process job 3The val qualifier can also be used to specify the process output. In this example, the Hello World! string is implicitly converted into a channel that is input to the FOO process. This process prints the input to a file named file.txt, and returns the same input value as the output.
process FOO {
input:
val x
output:
val x
script:
"""
echo $x > file.txt
"""
}
workflow {
out_ch = FOO("Hello world!")
out_ch.view()
}The output from FOO is assigned to out_ch, and its contents printed using the view() channel operator.
>>> nextflow run foo.nf
N E X T F L O W ~ version 23.04.1
Launching `foo.nf` [dreamy_turing] DSL2 - revision: 0d1a07970e
executor > local (1)
[a4/f710b3] process > FOO [100%] 1 of 1 ✔
Hello world!Paths
The path qualifier allows the handling of files inside a process. When a new instance of a process is executed, a new process execution director will be created just for that process. When the path qualifier is specified as the input, Nextflow will stage the file inside the process execution directory, allowing it to be accessed by the script using the specified name in the input declaration.
In this example, the reads channel is created from multiple .fq files inside training/nf-training/data/ggal, and input into process FOO. In the input declaration of the process, the file is referred to as sample.fastq.
The training/nf-training/data/ggal folder contains multiple .fq files, along with a .fa file. The wildcard *is used to match only .fq to be used as input.
>>> ls training/nf-training/data/ggal
gut_1.fq gut_2.fq liver_1.fq liver_2.fq lung_1.fq lung_2.fq transcriptome.faSave the following code block as foo.nf.
reads = Channel.fromPath('training/nf-training/data/ggal/*.fq')
process FOO {
debug true
input:
path 'sample.fastq'
script:
"""
ls sample.fastq
"""
}
workflow {
FOO(reads)
}When the script is ran, the FOO process is executed six times and will print the name of the file sample.fastq six times, since this is the name assigned in the input declaration.
>>> nextflow run foo.nf
N E X T F L O W ~ version 23.04.1
Launching `foo.nf` [nasty_lamport] DSL2 - revision: b214838b82
[78/a8a52d] process > FOO [100%] 6 of 6 ✔
sample.fastq
sample.fastq
sample.fastq
sample.fastq
sample.fastq
sample.fastqInside the process execution directory (ie. work/78/a8a52d...), the input file has been staged (symbolically linked) under the input declaration name. This allows the script to access the file within the execution directory via the declaration name.
>>> ll work/78/a8a52d...
sample.fastq -> /.../training/nf-training/data/ggal/liver_1.fqSimilarly, the path qualifier can also be used to specify one or more files that will be output by the process. In this example, the RANDOMNUM process creates a file results.txt containing a random number. Note that the Bash function is escaped with a back-slash character (ie. \$RANDOM).
process RANDOMNUM {
output:
path "*.txt"
script:
"""
echo \$RANDOM > result.txt
"""
}
workflow {
receiver_ch = RANDOMNUM()
receiver_ch.view()
}The output file is declared with the path qualifier, and specified using the wildcard * that will output all files with .txt extension. The output of the RANDOMNUM process is assigned to receiver_ch, which can be used for downstream processes.
>>> nextflow run foo.nf
N E X T F L O W ~ version 23.04.1
Launching `foo.nf` [nostalgic_cajal] DSL2 - revision: 9e260eead5
executor > local (1)
[76/7e8e36] process > RANDOMNUM [100%] 1 of 1 ✔
/...work/8c/792157d409524d06b89faf2c1e6d75/result.txtTuples
To define paired/grouped input and output information, the tuple qualifier can be used. The input and output declarations for tuples must be declared with a tuple qualifier followed by the definition of each element in the tuple.
In the example below, reads_ch is a channel created using the fromFilePairs channel factory, which automatically creates a tuple from file pairs.
reads_ch = Channel.fromFilePairs("training/nf-training/data/ggal/*_{1,2}.fq")
reads_ch.view()The created tuple consists of two elements – the first element is always the grouping key of the matching pair (based on similarities in the file name), and the second is a list of paths to each file.
[gut, [/.../training/nf-training/data/ggal/gut_1.fq, /.../training/nf-training/data/ggal/gut_2.fq]]
[liver, [/.../training/nf-training/data/ggal/liver_1.fq, /.../training/nf-training/data/ggal/liver_2.fq]]
[lung, [/.../training/nf-training/data/ggal/lung_1.fq, /.../training/nf-training/data/ggal/lung_2.fq]]To input a tuple into a process, the tuple qualifier must be used in the input block. Below, the first element of the tuple (ie. the grouping key) is declared with the val qualifier, and the second element of the tuple is declared with the path qualifier. The FOO process then prints the .fq file paths to a file called sample.txt, and returns it as a tuple containing the same grouping key, declared with val, and the output file created inside the process, declared with path.
process FOO {
input:
tuple val(sample_id), path(sample_id_paths)
output:
tuple val(sample_id), path('sample.txt')
script:
"""
echo $sample_id_paths > sample.txt
"""
}
workflow {
sample_ch = FOO(reads_ch)
sample_ch.view()
}Update foo.nf to the above, and run the script.
>>> nextflow run foo.nf
N E X T F L O W ~ version 23.04.1
Launching `test.nf` [sharp_becquerel] DSL2 - revision: cd652fc08b
executor > local (3)
[65/54124a] process > FOO (3) [100%] 3 of 3 ✔
[lung, /.../work/23/fe268295bab990a40b95b7091530b6/sample.txt]
[liver, /.../work/32/656b96a01a460f27fa207e85995ead/sample.txt]
[gut, /.../work/ae/3cfc7cf0748a598c5e2da750b6bac6/sample.txt]It’s worth noting that the FOO process is executed three times in parallel, so there’s no guarantee of a particular execution order. Therefore, if the script was ran again, the final result may be printed out in a different order:
>>> nextflow run foo.nf
N E X T F L O W ~ version 23.04.1
Launching `foo.nf` [high_mendel] DSL2 - revision: cd652fc08b
executor > local (3)
[82/71a961] process > FOO (1) [100%] 3 of 3 ✔
[gut, /.../work/ae/3cfc7cf0748a598c5e2da750b6bac6/sample.txt]
[lung, /.../work/23/fe268295bab990a40b95b7091530b6/sample.txt]
[liver, /.../work/32/656b96a01a460f27fa207e85995ead/sample.txt]Thus, if the output of a process is being used as an input into another process, the use of the tuple qualifier that contains metadata information is especially important to ensure the correct inputs are being used for downstream processes.
- The contents of value channels can be consumed an unlimited amount of times, wheres queue channels cannot
- Different channel factories can be used to read different input types
$characters need to be escaped with\when referencing Bash variables and functions, while Nextflow variables do not- The scripting language within a process can be altered by starting the script with the desired Shebang declaration
Creating an RNAseq Workflow
- Develop a Nextflow workflow
- Read data of different types into a Nextflow workflow
- Output Nextflow process results to a predefined directory
4.1.1. Define Workflow Parameters
Let’s create a Nextflow script rnaseq.nf for a RNA-seq workflow. The code begins with a shebang, which declares Nextflow as the interpreter.
#!/usr/bin/env nextflowOne way to define the workflow parameters is inside the Nextflow script.
params.reads = "/.../training/nf-training/data/ggal/*_{1,2}.fq"
params.transcriptome_file = "/.../training/nf-training/data/ggal/transcriptome.fa"
params.multiqc = "/.../training/nf-training/multiqc"
println "reads: $params.reads"Workflow parameters can be defined and accessed inside the Nextflow script by prepending the prefix params to a variable name, separated by a dot character, eg. params.reads.
Different data types can be assigned as a parameter in Nextflow. The reads parameter is defined as multiple .fq files. The transcriptome_file parameter is defined as one file, /.../training/nf-training/data/ggal/transcriptome.fa. The multiqc parameter is defined as a directory, /.../training/nf-training/data/ggal/multiqc.
The Groovy println command is then used to print the contents of the reads parameter, which is access with the $ character.
Run the script:
>>> nextflow run rnaseq.nf
N E X T F L O W ~ version 23.04.1
Launching `rnaseq.nf` [astonishing_raman] DSL2 - revision: 8c9adc1772
reads: /.../training/nf-training/data/ggal/*_{1,2}.fq4.1.2. Create a transcriptome index file
Commands or scripts can be executed inside a process.
process INDEX {
input:
path transcriptome
output:
path "salmon_idx"
script:
"""
salmon index --threads $task.cpus -t $transcriptome -i salmon_idx
"""
}The INDEX process takes an input path, and assigns that input as the variable transcriptome. The path type qualifier will allow Nextflow to stage the files in the process execution directory, where they can be accessed by the script via the defined variable name, ie. transcriptome. The code between the three double-quotes of the script block will be executed, and accesses the input transcriptome variable using $. The output is a path, with a filename salmon_idx. The output path can also be defined using wildcards, eg. path "*_idx".
Note that the name of the input file is not used and is only referenced to by the input variable name. This feature allows pipeline tasks to be self-contained and decoupled from the execution environment. As best practice, avoid referencing files that are not defined in the process script.
To execute the INDEX process, a workflow scope will need to be added.
workflow {
index_ch = INDEX(params.transcriptome_file)
}Here, the params.transcriptome_file parameter we defined earlier in the Nextflow script is used as an input into the INDEX process. The output of the process is assigned to the index_ch channel.
Run the Nextflow script:
>>> nextflow run rnaseq.nf
ERROR ~ Error executing process > 'INDEX'
Caused by:
Process `INDEX` terminated with an error exit status (127)
Command executed:
salmon index --threads 1 -t transcriptome.fa -i salmon_index
Command exit status:
127
Command output:
(empty)
Command error:
.command.sh: line 2: salmon: command not found
Work dir:
/.../work/85/495a21afcaaf5f94780aff6b2a964c
Tip: you can try to figure out what's wrong by changing to the process work dir and showing the script file named `.command.sh`
-- Check '.nextflow.log' file for detailsWhen a process execution exits with a non-zero exit status, the workflow will be stopped. Nextflow will output the cause of the error, the command that caused the error, the exit status, the standard output (if available), the comand standard error, and the work directory where the process was executed.
Let’s first look inside the process execution directory:
>>> ls -a /.../work/85/495a21afcaaf5f94780aff6b2a964c
. .command.begin .command.log .command.run .exitcode
.. .command.err .command.out .command.sh transcriptome.faWe can see that the input file transcriptome.fa has been staged inside this process execution directory by being symbolically linked. This allows it to be accessed by the script.
Inside the .command.err script, we can see that the salmon command was not found, resulting in the termination of the Nextflow workflow.
Singularity containers can be used to execute the process within an environment that contains the package of interest. Create a config file nextflow.config containing the following:
singularity {
enabled = true
autoMounts = true
cacheDir = "/config/binaries/singularity/containers_devel/nextflow"
}The container process directive can be used to specify the required container:
process INDEX {
container "/config/binaries/singularity/containers_devel/nextflow/depot.galaxyproject.org-singularity-salmon-1.10.1--h7e5ed60_0.img"
input:
path transcriptome
output:
path "salmon_idx"
script:
"""
salmon index --threads $task.cpus -t $transcriptome -i salmon_idx
"""
}Run the Nextflow script:
>>> nextflow run rnaseq.nf
N E X T F L O W ~ version 23.04.1
Launching `rnaseq.nf` [distraught_goldwasser] DSL2 - revision: bdebf34e16
executor > local (1)
[37/7ef8f0] process > INDEX [100%] 1 of 1 ✔The newly created nextflow.config files does not need to be specified in the nextflow run command. This file is automatically searched for and used by Nextflow.
An alternative to singularity containers is the use of a module. Since the script block is executed as a Bash script, it can contain any command or script normally executed on the command line. If there is a module present in the host environment, it can be loaded as part of the process script.
process INDEX {
input:
path transcriptome
output:
path "salmon_idx"
script:
"""
module purge
module load salmon/1.3.0
salmon index --threads $task.cpus -t $transcriptome -i salmon_idx
"""
}Run the Nextflow script:
>>> nextflow run rnaseq.nf
N E X T F L O W ~ version 23.04.1
Launching `rnaseq.nf` [reverent_liskov] DSL2 - revision: b74c22049d
executor > local (1)
[ba/3c12ab] process > INDEX [100%] 1 of 1 ✔4.1.3. Collect Read Files By Pairs
Previously, we have defined the reads parameter to be the following:
params.reads = "/.../training/nf-training/data/ggal/*_{1,2}.fq"Challenge: Convert the reads parameter into a tuple channel called reads_ch, where the first element is a unique grouping key, and the second element is the paired .fq files. Then, view the contents of reads_ch
reads_ch = Channel.fromFilePairs("$params.reads")
reads_ch.view()The fromFilePairs channel factory will automatically group input files into a tuple with a unique grouping key. The view() channel operator can be used to view the contents of the channel.
>>> nextflow run rnaseq.nf
[gut, [/.../training/nf-training/data/ggal/gut_1.fq, /.../training/nf-training/data/ggal/gut_2.fq]]
[liver, [/.../training/nf-training/data/ggal/liver_1.fq, /.../training/nf-training/data/ggal/liver_2.fq]]
[lung, [/.../training/nf-training/data/ggal/lung_1.fq, /.../training/nf-training/data/ggal/lung_2.fq]]4.1.4. Perform Expression Quantification
Let’s add a new process QUANTIFICATION that uses both the indexed transcriptome file and the .fq file pairs to execute the salmon quant command.
process QUANTIFICATION {
input:
path salmon_index
tuple val(sample_id), path(reads)
output:
path "$sample_id"
script:
"""
salmon quant --threads $task.cpus --libType=U \
-i $salmon_index -1 ${reads[0]} -2 ${reads[1]} -o $sample_id
"""
}The QUANTIFICATION process takes two inputs, the first is the path to the salmon_index created from the INDEX process. The second input is set to match the output of fromFilePairs – a tuple where the first element is a value (ie. grouping key), and the second element is a list of paths to the .fq reads.
In the script block, the salmon quant command saves the output of the tool as $sample_id. This output is emitted by the QUANTIFICATION process, using $ to access the Nextflow variable.
Challenge:
Set the following as the execution container for QUANTIFICATION:
/config/binaries/singularity/containers_devel/nextflow/depot.galaxyproject.org-singularity-salmon-1.10.1--h7e5ed60_0.imgAssign index_ch and reads_ch as the inputs to this process, and emit the process outputs as quant_ch. View the contents of quant_ch
To assign a container to a process, the container directive can be used.
process QUANTIFICATION {
container "/config/binaries/singularity/containers_devel/nextflow/depot.galaxyproject.org-singularity-salmon-1.10.1--h7e5ed60_0.img"
input:
path salmon_index
tuple val(sample_id), path(reads)
output:
path "$sample_id"
script:
"""
salmon quant --threads $task.cpus --libType=U \
-i $salmon_index -1 ${reads[0]} -2 ${reads[1]} -o $sample_id
"""
}To run the QUANTIFICATION process and emit the outputs as quant_ch, the following can be added to the end of the workflow block:
quant_ch = QUANTIFICATION(index_ch, reads_ch)
quant_ch.view()The script can now be run:
>>> nextflow run rnaseq.nf
N E X T F L O W ~ version 23.04.1
Launching `rnaseq.nf` [elated_cray] DSL2 - revision: abe41f4f69
executor > local (4)
[e5/e75095] process > INDEX [100%] 1 of 1 ✔
[4c/68a000] process > QUANTIFICATION (1) [100%] 3 of 3 ✔
/.../work/b1/d861d26d4d36864a17d2cec8d67c80/liver
/.../work/b4/a6545471c1f949b2723d43a9cce05f/lung
/.../work/4c/68a000f7c6503e8ae1fe4d0d3c93d8/gutIn the Nextflow output, we can see that the QUANTIFICATION process has been ran three times, since the reads_ch consists of three elements. Nextflow will automatically run the QUANTIFICATION process on each of the elements in the input channel, creating separate process execution work directories for each execution.
4.1.5. Quality Control
Now, let’s implement a FASTQC quality control process for the input fastq reads.
Challenge:
Create a process called FASTQC that takes reads_ch as an input, and declares the process input to be a tuple matching the structure of reads_ch, where the first element is assigned the variable sample_id, and the second variable is assigned the variable reads. This FASTQC process will first create an output directory fastqc_${sample_id}_logs, then perform fastqc on the input reads and save the results in the newly created directory fastqc_${sample_id}_logs:
mkdir fastqc_${sample_id}_logs
fastqc -o fastqc_${sample_id}_logs -f fastq -q ${reads}Take fastqc_${sample_id}_logs as the output of the process, and assign it to the channel fastqc_ch. Finally, specify the process container to be the following:
/config/binaries/singularity/containers_devel/nextflow/depot.galaxyproject.org-singularity-fastqc-0.12.1--hdfd78af_0.imgThe process FASTQC is created in rnaseq.nf. Since the input channel is a tuple, the process input declaration is a tuple containing elements that match the structure of the incoming channel. The first element of the tuple is assigned the variable sample_id, and the second element of the tuple is assigned the variable reads. The relevant container is specified using the container process directive.
process FASTQC {
container "/config/binaries/singularity/containers_devel/nextflow/depot.galaxyproject.org-singularity-fastqc-0.12.1--hdfd78af_0.img"
input:
tuple val(sample_id), path(reads)
output:
path "fastqc_${sample_id}_logs"
script:
"""
mkdir fastqc_${sample_id}_logs
fastqc -o fastqc_${sample_id}_logs -f fastq -q ${reads}
"""
}In the workflow scope, the following can be added:
fastqc_ch = FASTQC(reads_ch)The FASTQC process is called, taking reads_ch as an input. The output of the process is assigned to be fastqc_ch.
>>> nextflow run rnaseq.nf
N E X T F L O W ~ version 23.04.1
Launching `rnaseq.nf` [sad_jennings] DSL2 - revision: cfae7ccc0e
executor > local (7)
[b5/6bece3] process > INDEX [100%] 1 of 1 ✔
[32/46f20b] process > QUANTIFICATION (3) [100%] 3 of 3 ✔
[44/27aa8d] process > FASTQC (2) [100%] 3 of 3 ✔In the Nextflow output, we can see that the FASTQC has been ran three times as expected, since the reads_ch consists of three elements.
4.1.6. MultiQC Report
So far, the generated outputs have all been saved inside the Nextflow work directory. For the FASTQC process, the specified output directory is only created inside the process execution directory. To save results to a specified folder, the publishDir process directive can be used.
Let’s create a new MULTIQC process in our workflow that takes the outputs from the QUANTIFICATION and FASTQC processes to create a final report using the multiqc tool, and publish the process outputs to a directory outside of the process execution directory.
process MULTIQC {
publishDir params.outdir, mode:'copy'
container "/config/binaries/singularity/containers_devel/nextflow/depot.galaxyproject.org-singularity-multiqc-1.21--pyhdfd78af_0.img"
input:
path quantification
path fastqc
output:
path "*.html"
script:
"""
multiqc . --filename $quantification
"""
}In the MULTIQC process, the multiqc command is performed on both quantification and fastqc inputs, and publishes the report to a directory defined by the outdir parameter. Only files that match the declaration in the output block are published, not all the outputs of a process. By default, files are published to the target folder creating a symbolic link to the file produced in the process execution directory. This behavior can be modified using the mode option, eg. copy, which copies the file from the process execution directory to the specified output directory.
Add the following to the end of workflow scope:
multiqc_ch = MULTIQC(quant_ch, fastqc_ch)Run the pipeline, specifying an output directory using the outdir parameter:
nextflow run rnaseq.nf --outdir "results"A results directory containing the output multiqc reports will be created outside of the process execution directory.
>>> ls results
gut.html liver.html lung.html- Commands or scripts can be executed inside a process
- Environments can be defined using the
containerprocess directive - The input declaration for a process must match the structure of the channel that is being passed into that process
This workshop is adapted from Fundamentals Training, Advanced Training, Developer Tutorials, and Nextflow Patterns materials from Nextflow and nf-core